Excited states of molecules. Rydberg spectra, predissociation, autoionization
Excited electronic states of molecules are very important for the chemistry of the atmosphere, the chemistry of the interstellar medium, energy storage, spectroscopy, laser systems, heterogeneous catalysis and the chemistry of molecules on metal and semiconductor surfaces.
Theoretical treatment of the excited states even for simple diatomic molecules has quite different requirements from ground-state treatments. In general, there are excited states of different character, such as valence, Rydberg, charge-transfer etc., and many types of interactions, such as radial, rotational-electronic, spin-orbit etc. that couple them. Multi-reference, large-scale configuration interaction calculations are required for the proper treatment of excited states of molecules, within the Born Oppenheimer approximation, followed by treatments of the non-adiabatic interactions.
Polyatomic systems pose the added challenge of identifying the different electronic states characterized by different point group symmetry at different molecular geometry, eg. the NO2 molecule.
Dissociative recombination of molecular cations requires determination of potential energy curves over the whole range of internuclear distances from the molecular region out to dissociation and for excited states up to the ionic limits, including states above the lowest ionization threshold, eg. CF/CF+
In the TPCI, we employ the Multi Reference configuration interaction method (MRDCI) http://www2.uni-wuppertal.de/FB9/praha/buenker/buenker.html for the basic electronic structure calculations and coupling matrix elements. Subsequently, calculations are carried out for
. Radiative transitions, bound-bound and radiative dissociation.
. Application of the complex scaling method for predissociation resonances in multi-state problems and electronic autoionization resonances in diatomic molecules.
. Prediction of the highly excited Rydberg states, spectral perturbations and vibrational autoionization by the MQDT method in diatomic and triatomic molecules.
At TPCI we have systematically studied by the above methods systems such as the Rydberg molecules and rare-gas dimers.
References to current work
1. I. D. Petsalakis and G. Theodorakopoulos "Theoretical calculations on the potential energy curves of electronic states of CF. Rydberg states of CF above the lowest ionization limit" Chem.Phys.Lett. 508, 17 (2011).
2. D. Irimia, I. D. Petsalakis, G. Theodorakopoulos and M. H.M. Janssen, Coherent oscillatory femtosecond dynamics in multichannel photodynamics of NO2 studied by spatially masked electron imaging, J. Phys. Chem. A 114, 3157 (2010)
3. I. D. Petsalakis, G. Theodorakopoulos ,, A. Grochol, P. Kowalczyk, W. Jastrzebski, Theoretical study of highly excited 1Sigma+ and 1P states of NaLi and experimental observation of the interacting 5 1Sigma+ and 6 1Sigma+ states, Chem. Phys. 362, 130 (2009)
4. I .D. Petsalakis, D. Tzeli and G. Theodorakopoulos, Theoretical study on the electronic states of NaLi. J. Chem. Phys. 129, 054306 (2008).
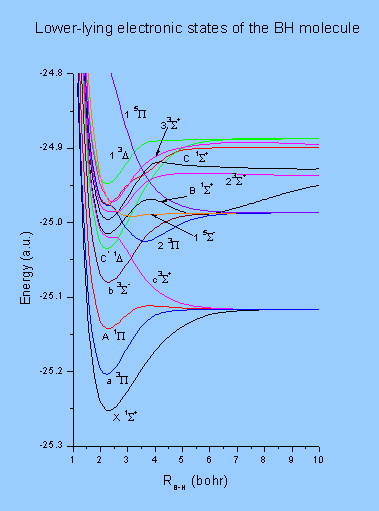
5. I. D. Petsalakis and G. Theodorakopoulos, Theoretical study of nonadiabatic interactions, radiative lifetimes and predissociation lifetimes of excited states of BH. Molecular Physics 105 333 (2007)
6. I. D. Petsalakis and G. Theodorakopoulos, Multireference configuration interaction and quantum defect calculations on the Rydberg states of the BH molecule. Mol. Phys. 104,103 (2006)
7. G. Theodorakopoulos and I. D. Petsalakis, Theoretical Study on the low-lying electronic states of InN. Chem. Phys. Lett. 423, 445 (2006)