Functionals of the one-body reduced density matrix
Although an old idea, introduced in the early days of quantum chemistry, reduced density matrix functional theory (RDMFT) has been explored and applied to real systems only the last two decades. In RDMFT, the one-body reduced density matrix (1-RDM) is the fundamental variable in analogy to density functional theory (DFT) where this role is played by the electronic density. The main task, in RDMFT, is to construct approximate expressions (functionals) of the total energy in terms of the 1-RDM and minimize them in order to obtain the total energy as well as the one-body reduced density matrix of the ground state. The main target of RDMFT is not to replace well established theories of the electronic structure but to complement them and improve over their results for the systems and properties that these theories do not describe properly. Such systems are, for example, those collectively termed as "highly correlated systems".
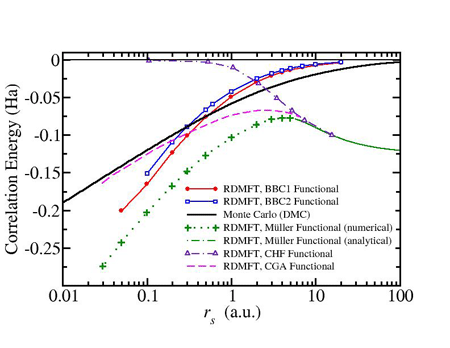
Our aim is to contribute to the development and assessment of RDMFT approximate functionals. We applied existing functionals of RDMFT to prototype systems like the homogeneous electron gas [1] (Fig. 1). Apart from assessment purposes, this application serves as a laboratory for the development of more accurate approximations [2].
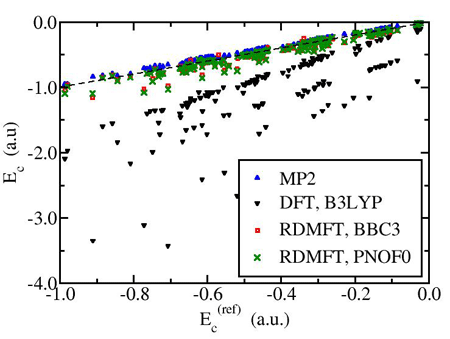
Additionally, we attempt to construct approximations by employing forms with physical meaning and fitting parameters to reproduce known properties of small molecular systems. Two such functional, we developed, is the ML [3] and Power functionals [4]. We performed large scale evaluation tests for the most important functionals for quantities like correlation and atomization energies for a large set of (~150) molecules and radicals (fig. 2)[5]. We have created a computer code implementing all functionals, which can provide full comparative assessment of all approximations for finite systems based on a Gaussian-type-basis expansion of the molecular orbitals.
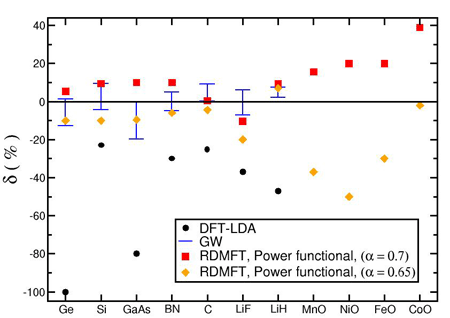
We applied 1-RDM functionals to periodic systems with focus on the famous band gap problem which is one of the pathologies of most DFT approximations. A recipe for the calculation of the fundamental gap is used, which is based on Energy-derivative discontinuities [6]. We showed that there are RDMFT functionals that reproduce the fundamental gaps of semiconductors and insulators [7] including those with Mott insulator behavior (fig. 3).
We studied quality features of functionals like size consistency [8] and the behavior in the regime of fractional spins [9]. Finally, a recently defined single electron spectrum given by a recipe similar to Janak's theorem in DFT was applied in the study of ionization energies and electron affinities of molecular and ionic systems [10].
The target of this research direction is to contribute in the development of theoretical tools based on RDMFT for the accurate description of systems, like highly correlating materials, for which the results of most density functional approximations are not satisfactory.
Fig. 1:The correlation energy of the Homogeneous Electron Gas (Jelium) using different RDMFT functionals compared to Monte-Carlo results.
Fig. 2:Plot of the correlation energy calculated with various methods versus the exact for a set of 150 molecules and radicals. The dashed line corresponds to perfect agreement with the exact.
Fig. 3: Error in the calculated gaps for DFT-LDA, GW method and RDMFT power-functional.