The motion of the electrons around nuclei is described via Schroedinger's wave function. For many electrons Schroedinger's equation cannot be solved exactly. There are two ways to solve it approximately: (i) by direct numerical integration, possible only for single atoms and diatomic molecules, and (ii) in terms of functions, whose integrals are known analytically, possible for larger molecules as well.
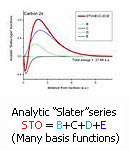
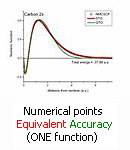
Carbon 2s orbital
The numerically obtained wave function of case (i) is conceptually simple, tailored to the problem, but is given as a set of points: It is not analytic, not always convenient for applications.
In case (ii) the usual choice of analytic functions of "Gauss" or "Slater" type demands too many of them to achieve the same accuracy: The result is a complicated wave function, also not always convenient for applications.
Q. : Is it possible to obtain handy analytic functions of "numerical" accuracy?
A. : Yes, by varying the shape of appropriate simple functions "tailored" to the problem at hand.
At TPCI we developed such functions for atoms and diatomic molecules within the standard method of "Configuration Interaction", one of the most accurate computational techniques available.
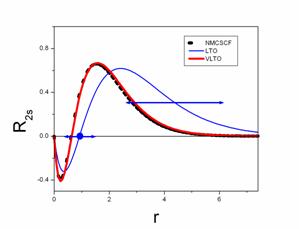
For atoms, the shape of the standard hydrogenic Laguerre functions (LTO) is varied by changing the node position and the spatial extent to minimize the total energy.
The coefficients a, b, c, . Z, are varied by keeping simultaneous orthogonality to lower energy functions, producing the "Variational Laguerre-type" functions (VLTO).
Thus, e.g. for Carbon, 13 VLTOs and 64 configurations obtain comparable accuracy with 145 "Gauss" functions and 1.500.000 configurations.
Similar Analytic functions have been developed for diatomic molecules too, by varying the shape of the standard One-Electron Diatomic Molecule (OEDM) functions.
These simple analytic functions have accuracy equivalent to the ''numerical'' solution and are potentially handier for applications.