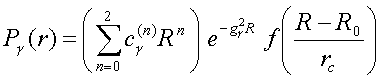Electronic structure of clusters from selected ab initio data
Clusters of atoms (species in between small molecules and solids) have recently become experimentally manageable to controllable sizes. This increases their usefulness in a large variety of chemical & microelectronic applications, provided the details of their electronic structure are known. Accurate ab-initio computations of the electronic structure for any desirable cluster currently place excessive demands on computing time.
Q. : Using information from a limited number and type of clusters is it possible to make fast and accurate predictions of properties of similar clusters of different sizes and shapes?
A. : Yes, using the
The Spin-Polarized Tight-Binding Interpolating Method.
At TPCI we extended the Tight-Binding-Method, developed at the Center for Computational Materials Science of the Naval Research Laboratory (USA), to include spin polarization , necessary for magnetic solids, as well as for atomic clusters and molecules. A selected database of accurate large-scale ab-initio electronic structure calculations, has been fitted in terms of instantaneously computed parametric analytic angle- and bond-length- dependent Hamiltonian matrix elements; this enables us to describe by interpolation - in a natural fashion - properties of clusters of different geometries and sizes of the same material, bypassing new lengthy ab-initio calculations.
For example the bond-length (R) - dependence of the TB Hamiltonian hopping matrix elements, in terms of the NRL parameters (c γ(n) and g γ below), can be expressed as a function of the form:
where γ represents ss σ , sp σ , . dd δ bonds, and f is a cutoff function.
Once the parameters c γ(n) and g γ are determined by fitting to a selected database of ab initio results, desired properties of other clusters, can be computed instantaneously.
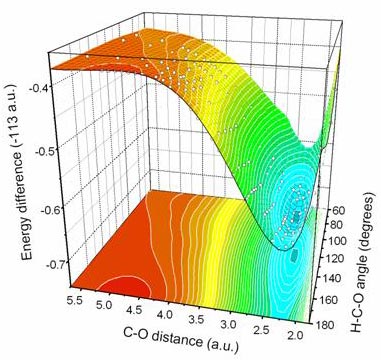
Potential Energy Surface of HCO from selected ab-initio data
The Spin-Polarized TB Method has been successfully tested in a prototype small molecule, HCO, by fitting to a database of highly accurate ab initio calculated energy values of 500 selected HCO geometries.
The figure displays a prediction of the Potential Energy Surface (PES) of HCO, based on an ab initio database (white points) calculated with the highly accurate method of Configuration Interaction (CI). The points are accurately fitted by the parametric Tight - Binding Hamiltonian.