The Optimized Effective Potential (OEP) method is one of the ways that can improve accuracy in Density Functional Theory. Within this method, explicit orbital functionals are minimized in terms of the local potential. This is an equivalent procedure to minimization with respect to the electron density.
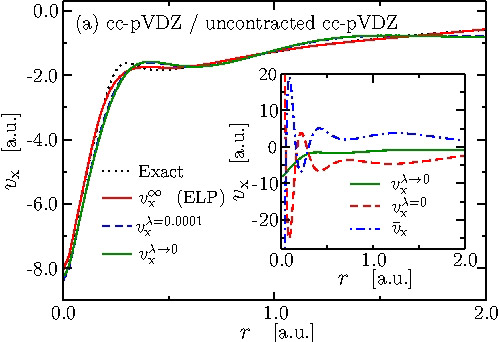
When the potential and the orbitals are expanded in finite size basis sets, the OEP method is well-known to lead to an ill-posed problem with a non-unique solution for the potential. We showed that the underlying mathematical problem is the discontinuous behavior of the solution of the OEP equation (Fredholm equation of the first kind) when the density-density response function (i.e. the kernel of the integral equation) is truncated with a finite orbital basis set and then inverted. We proposed a remedy for this problem that leads to a sound definition of OEP for finite basis sets (Fig. 1).
Fig. 1:Effective potential for Ne atom taking the correct limit when basis set is truncated.
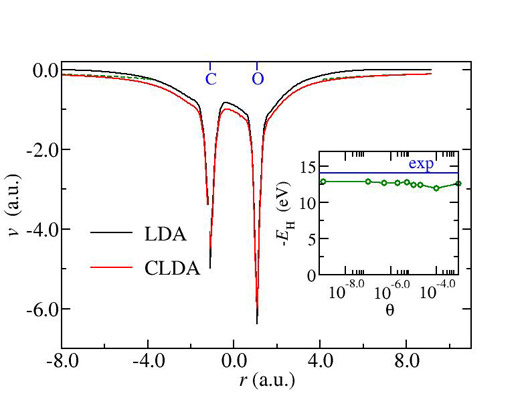
Self Interactions (SI) is a major problem in most Density Functional Approximations. We proposed a remedy which is based on the minimization of any DFA total energy in terms of the local potential in the OEP fashion, under two subsidiary conditions. The first of these conditions restricts the optimal potential to have the correct asymptotic behavior (N-1)/r. The second, restricts the corresponding “screening-density” defined as the negative Laplacian of the potential to be positive in all space. Under these conditions we demonstrate that a major manifestation of SI in LDA, namely the severe underestimation of Ionization energies and electron affinities is largely corrected (Fig. 2).
Fig 2.: The LDA potential for CO molecule compared with the constrained OEP-LDA potential with improved asymptotic behavior.
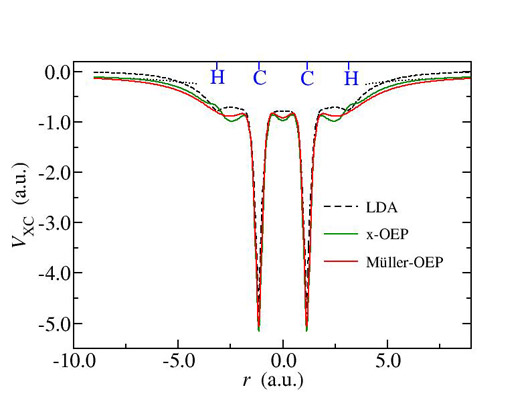
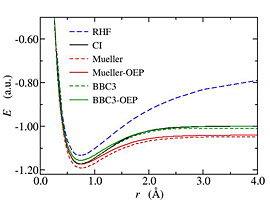
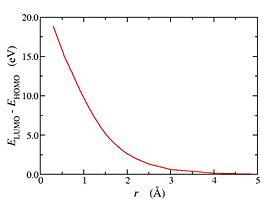
The OEP method can be used in reduced density matrix functional theory (RDMFT) as an approximate but very efficient way to minimize functionals with respect to the approximate natural orbitals. On the other hand RDMFT functionals can be viewed as correlated functionals in OEP theory beyond the exact exchange. Finally, the spectrum of the effective local potential offers a single electron spectrum missing from RDMFT. We implemented OEP theory for RDMFT (Fig. 3) and showed that in most cases the quality of the functional in reproducing electronic correlations is not affected by the restriction of local effective potential yielding approximate natural orbitals (Fig. 4). In addition we demonstrate the usefulness of the single electron spectrum of the local potential by comparing the HOMO energies with ionization energies.
Fig. 3: Local potentials for the acetylene molecule using exact exchange (x) OEP and OEP- Mueller RDMFT functional compared to the LDA local potential.
Fig. 4:Dissociation of H2 molecule using plain RDMFT functionals (Mueller and BBC3) and their OEP-RDMFT version (left). The correct dissociation offered by RDMFT functionals is not impaired by using OEP. In addition OEP-RDMFT reproduces the HOMO-LUMO degeneracy at dissociation (right).